
Síndrome de Marfan
- O que é a patologia?
A Síndrome de Marfan (SM) é uma doença hereditária e multissistêmica47 do tecido conjuntivo, com transmissão autossômica dominante.1,4,5 Apresenta uma incidência de um caso por cada 5000 indivíduos e não demonstra predileção47 geográfica, étnica ou de gênero. Também é conhecida como aracnodactilia, pois é um dos sinais da síndrome de Marfan, que se caracteriza por dedos das mãos e pés anormalmente longos, delgados47 ou finos e a magreza também se torna um fator recorrente.6 Ocorre por causa de uma mutação do gene FBN1, no cromossomo 15, responsável pela produção da fibrilina. Essa proteína é necessária para fornecer elasticidade e apoio aos tecidos conjuntivos.2 As principais manifestações envolvem os sistemas cardiovascular (aneurisma da aorta proximal e dissecção aórtica47), ocular (luxação47 da lente ocular) e esquelético, embora também possa ocorrer envolvimento do pulmão (pneumotórax espontâneo e bolhas apicais47), pele (estria atrófica e hérnia47 recorrente) e sistema nervoso central.3,8 Muitas características da Síndrome de Marfan progridem ao longo do tempo.8 A diminuição da expectativa de vida ocorre principalmente devido a complicações aórticas.30
- Parte biológica
A fibrilina-1, uma glicoproteína47 ligante do cálcio rica em cisteína, é o principal componente das microfibrilas extracelulares (Figura 1). Ela é organizada em fibras multiméricas compostas de dezenas ou centenas de monômeros47 que são sintetizados por várias células da matriz extracelular, incluindo fibroblastos, osteoblastos, células de músculo liso e pericondrócitos. Suas funções estão diretamente relacionadas às das microfibrilas, já que são o seu principal componente. As microfibrilas atuam como um suporte para a deposição de tropoelastina, determinam a direção de crescimento das fibras elásticas e desempenham um papel importante na ligação entre as fibras elásticas umas às outras, entre as fibras elásticas e outros componentes da matriz extracelular e entre as fibras elásticas e as células. As microfibrilas de fibrilina são elementos arquitetônicos distintos que são onipresentes no espaço do tecido conjuntivo e também únicos, exibindo padrões específicos do tecido.30 Pesquisas recentes mostraram que a fibrilina-1 também tem outras funções, incluindo a ligação do fator de crescimento TGF-β (fator de crescimento transformador beta) na matriz extracelular. A falta de fibrilina-1, funcionando normalmente, leva a uma liberação descontrolada de TGF-β e quando se liga aos receptores de TGF-β, a via de sinalização de TGF-β é ativada para que o crescimento aumente. A fibrilina-1 está distribuída tanto no tecido conjuntivo elástico quanto no inelástico, ao longo de todo o corpo, exceto no vítreo47 e na retina. Alterações no gene da fibrilina-1 levam à Síndrome de Marfan. Essa patologia é extremamente agressiva, nos órgãos que são afetados pela mutação14, suas características cardinais são recorrentes em 3 sistemas - esquelético, ocular e cardiovascular.12
Figura 1 - Processo de formação de microfibrilas a partir da fibrilina-1 e o mecanismo de ligação da molécula de TGF-β à fibrilina-1

Fonte: BYERS, 2004 adaptado por GYURICZA.45
Sistema esquelético: A característica mais marcante desta síndrome rara é o aumento do comprimento dos ossos longos, especialmente das mãos e dos pés, a aracnodactilia, identificada pelos dedos longos e finos. Os pacientes são tipicamente altos e magros, provocando a dolicostenomelia, caracterizada pela desproporção entre a relação da envergadura e altura (razão da envergadura dos braços × altura > 1,05; razão do segmento superior para inferior < 0,85 em adultos).7,19 Essas manifestações esqueléticas, que consistem na elevação da estatura, alterações da caixa torácica e curvaturas espinhais severas, podem desencadear deformações, como a escoliose presente em aproximadamente 62% dos pacientes, o crescimento excessivo das costelas pode fazer com que o osso do peito (esterno) se curve para dentro (pectus excavatum ou tórax em funil) ou empurre para fora (pectus carinatum ou peito de pombo) (Figura 2), em 60% deles.7,26 Isso significa que o corpo tem menos absorção e suspensão de choque.19 A protrusão acetabular é geralmente assintomática em adultos jovens. Os pés planos estão frequentemente presentes, enquanto alguns doentes apresentam pés cavos. A camptodactilia é frequentemente observada, especialmente nas crianças com grave e rápida progressão da doença. Várias características faciais estão tipicamente presentes: dolicocefalia, palato ogival47, apinhamento dentário, retrognatismo47 ou micrognatia, osso malar achatado e fissuras palpebrais inclinadas para baixo.7
Figura 2 - Uma pessoa com pectus carinatum, no qual o esterno é empurrado para fora.

Fonte: GeneReviews, © 1993-2020 University of Washington37
Algumas anomalias esqueléticas da SM são comuns na população geral como, por exemplo, a laxidez47 articular. Neste contexto, só a presença de uma combinação de alterações permite aumentar a especificidade diagnóstica. A radiologia é útil para a detecção de algumas anomalias.
A Ectasia Dural47 é o crescimento da dura e é comum em desordens de tecido conectivo20, definida pelo alargamento do canal raquidiano ao nível da região lombo-sagrada, está presente em 62 a 93% dos doentes, estando a sua identificação dependente da realização de tomografia computorizada ou de ressonância magnética. A associação desta alteração a queixas álgicas lombares permanece controversa.7
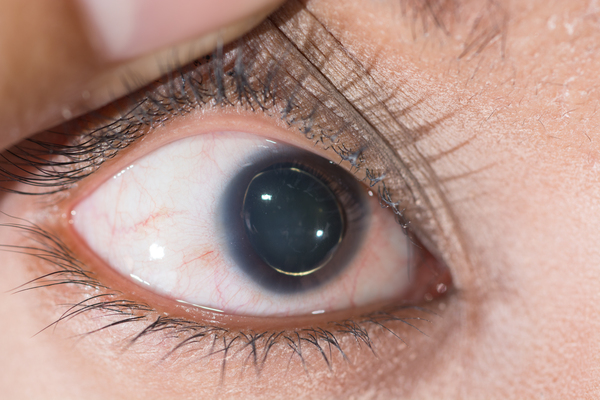
Sistema ocular: A visão de quem apresenta a síndrome de Marfan, possui problemas por causa da fibrilina. A miopia é o problema mais comum, e se deve em parte ao fato de que o olho cresceu mais do que o normal e se tornou mais longo (aumento do comprimento axial do globo ocular), por isso, devido à curva aumentada da retina com a mudanças no tecido conjuntivo no globo ocular, existe o desenvolvimento de dificuldades para a visualização de objetos que estão a longa distância, além de deslocamento (luxação) do cristalino do centro da pupila (ectopia lentis) (Figura 3).12,19 Indivíduos com Síndrome de Marfan também apresentam risco aumentado de descolamento de retina, glaucoma, formação precoce de catarata6, córnea achatada e hipoplasia da íris. No caso de deficiência visual permanente, o contato com uma unidade de visão pode ser necessário para testar os recursos visuais. Além disso, um estudo revelou que de 573 pacientes com Síndrome de Marfan , foi descoberto que 110 (19,2%) tinham estrabismo47; a exotropia ocorreu em 67 indivíduos (11,7%), a esotropia em 12 (2,1%), os desvios verticais em 8 pacientes (1,4%) e a hiperatividade do músculo oblíquo inferior primário, em 3 portadores de Marfan (0,5%).21
Figura 3 - Ectopia lentis

Fonte: ARZTSAMUI / Shutterstock.com38
Sistema cardiovascular: Os pacientes com esta síndrome estão predispostos a complicações cardíacas, é frequente o espessamento e/ou prolapso das válvulas auriculoventriculares47, uma ocorrência tardia e interpretada como sendo secundária à dilatação da raiz da aorta (principal manifestação cardiovascular). Devido à degeneração cística da camada média da aorta, essas dissecções normalmente começam acima dos óstios coronários e podem estender-se por toda a extensão da aorta, provocando insuficiência na válvula mitral, que pode ser complicada por arritmias (potencial causa de morte súbita em pacientes com SM), endocardite ou insuficiência cardíaca.18
O diâmetro aórtico, medido ao nível dos seios de Valsalva47, pode ser avaliado por ecocardiograma, angioTC ou angioressonância e o valor obtido deve ser indexado à superfície corporal e à idade do doente. Dos exames mencionados, embora o ecocardiograma seja o mais utilizado, é também o menos preciso e deve ter-se em conta que medições oblíquas subestimam o diâmetro aórtico. Após uma avaliação imagiológica basal, esta deve ser repetida aos 6 meses, para quantificar a taxa de progressão da dilatação, e se estável, a avaliação deverá passar a ser realizada anualmente. As alterações identificadas nas paredes aneurismáticas da aorta de doentes com SM, incluem: fragmentação das fibras elásticas (elastólise) com desorganização da lâmina elástica; degenerescência cística da média, com acumulação de glicosaminoglicanos; apoptose das células de músculo liso vascular (CMLV); aumento da expressão de metaloproteinases da matriz extracelular; ausência de infiltrado inflamatório. Ocorre uma diminuição das ligações entre a lâmina elástica e as CMLV e em condições de sobrecarga hemodinâmica pode exacerbar-se esta anomalia, registrando-se um aumento da produção de elementos da MEC.7
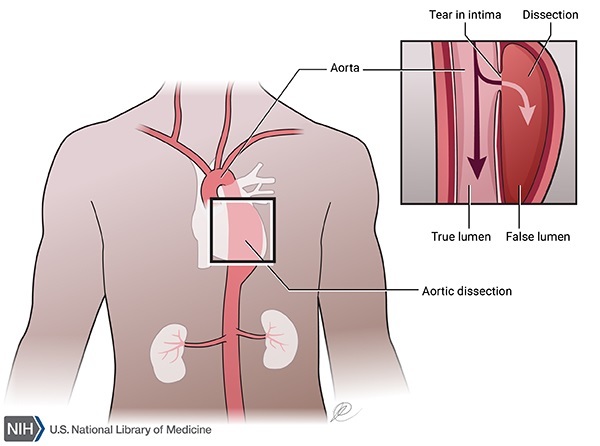
Há constatação que a dissecção da aorta (Figura 4) é extremamente rara em crianças, independentemente das dimensões do vaso. Nos adultos, os dois principais fatores de risco para dissecção aórtica são o diâmetro máximo da aorta e a presença de história familiar de dissecção aórtica. O risco de complicações aórticas (ruptura e dissecção) é significativamente maior quando o diâmetro da raiz da aorta é superior a 5 cm, motivo pelo qual está atualmente indicada a realização de uma cirurgia eletiva de substituição simultânea da raiz e válvula aórtica (cirurgia de Bental) a partir destes valores. Mulheres com SM apresentam um maior risco de dissecção aórtica durante a gravidez, sobretudo se tiverem um diâmetro da raiz da aorta superior a 4 cm no início da gravidez ou se houver progressão da dilatação ao longo do período gestacional. Os bloqueadores- μ não devem ser descontinuados neste período e a gravidez deve ser planejada e acompanhada de perto por especialistas em obstetrícia e cardiologia.7 As crianças com um início precoce e grave da SM apresentam frequentemente prolapso da válvula mitral com regurgitação valvular, conduzindo a dilatação e compromisso da função ventricular esquerda e hipertensão pulmonar.
Figura 4 - Dissecção da aorta

Fonte: Editado por Vitória Gomes da Silva Disponível na US National Library of Medicine39
Pulmões: o tecido elástico dos pulmões é alterado pela síndrome, a presença de maior distensibilidade é um fator de bom prognóstico. Os alvéolos podem ficar maiores e menores (enfisema), o que aumenta o risco de ruptura. A probabilidade de desenvolver enfisema aumenta se o tecido pulmonar for exposto a estresse prejudicial, por exemplo, através do fumo.12 O colapso pulmonar espontâneo, pneumotórax, ocorre em cerca de cinco por cento das pessoas que têm a síndrome, geralmente devido a uma ruptura de alguns alvéolos. O saco pulmonar é então preenchido com ar, ao mesmo tempo que o pulmão entra em colapso. Isso pode causar falta de ar e dor no peito e requer atendimento imediato no hospital, mesmo se a condição não for fatal.22 O ar é sugado para fora dos alvéolos e o paciente está completamente restabelecido. Alguns podem ter pneumotórax recorrente e, então, pode ser relevante uma operação para fins preventivos.
O envolvimento pulmonar pode resultar das alterações esqueléticas, como da deformação grave do esterno ou da coluna vertebral, que podem condicionar um padrão ventilatório restritivo. As alterações do parênquima incluem o desenvolvimento de bolhas (apicais).7
Outros sistemas: em doentes com SM, ao contrário de outras doenças do tecido conjuntivo, as lesões cutâneas não são usuais. A manifestação mais comum, que afeta cerca de dois terços dos doentes, é o aparecimento de estrias atróficas em locais não associados a distensão cutânea, como nos ombros, e de hérnias inguinais, tanto na altura do nascimento como na adolescência, com risco elevado de recorrência após cirurgia.1,7 Já nas articulações da mandíbula, se houver problemas, o tratamento fisiológico da mordida pode ser necessário.
Em caso de apneia do sono e problemas com ronco o paciente deve ser acompanhado por um médico. O apoio psicológico deve ser oferecido no momento do diagnóstico tanto para o paciente quanto para familiares e também continuamente de acordo com a necessidade.22
Diagnóstico
A síndrome de Marfan foi descrita pela primeira vez há mais de 100 anos por um professor parisiense de pediatria, Antoine-Bernard Marfan, que relatou a associação de dedos longos e delgados e outras anormalidades esqueléticas em uma menina de 5 anos, Gabrielle. Avanços importantes foram feitos no diagnóstico dos indivíduos afetados, embora morbidade substancial e mortalidade prematura permanecem associadas a esse transtorno.3
Para que o diagnóstico seja feito de forma exata, é necessário a atuação multiprofissional no paciente, como oftalmologistas, pediatras, neuropediatra e a cardiologia, além de reumatologista e geneticista.16 O diagnóstico da Síndrome de Marfan é estabelecido de acordo com uma revisão dos critérios diagnósticos, conhecida como nosologia de Ghent6,17(Tabela 1), por meio de uma avaliação abrangente amplamente baseada na combinação de manifestações clínicas maiores e menores em vários sistemas orgânicos e na história familiar.18 É importante realçar, que estes critérios não se aplicam a menores de 18 anos, dada a frequente expressão incompleta da doença nas crianças.7 Existem quatro principais características de diagnóstico clínico:12
- Dilatação ou dissecção da aorta ao nível dos seios da face de valsalva.
- Ectopia lentis (lente deslocada do olho).
- Ectasia dural lombossacra determinada por tomografia computadorizada ou ressonância magnética (MRI).
- Quatro das oito características esqueléticas típicas.
A incidência da doença é esporádica, os sintomas podem variar entre pacientes de uma mesma família, por tal motivo, se faz necessário a realização de múltiplos exames para fechamento do diagnóstico da síndrome de Marfan. De acordo com a necessidade visualizada no sujeito, o médico fará a solicitação de um ecocardiograma e uma tomografia computadorizada, além de uma ressonância magnética, teste de pressão ocular, testes genéticos, dentre outras solicitações que são importantes para que se possa descartar outras condições anormais, que tenham sintomas semelhantes. Embora não exista uma cura, a condição médica do tratamento concentra-se especialmente na prevenção de possíveis complicações.
Quadro 1 - Nosologia de Ghent46

- Base genética da herança
Cada gene que ocupa o mesmo lugar é chamado de alelo, quando ocorre uma mutação em apenas um dos alelos47, surgem os sintomas de uma doença autossômica dominante, como a Síndrome de Marfan.35 O gene FBN1 (Figura 5), localizado no cromossomo 15q21.1 (braço longo do cromossomo), é composto de 65 exons, codifica uma glicoproteína, a profibrilina-1, com cerca de 350KDa e que após processamento, se designa por fibrilina-1. A região promotora é grande e mal caracterizada. A alta conservação evolutiva da sequência intrônica na extremidade 5' do gene sugere a presença de elementos reguladores intrônicos. Três exons47 na extremidade 5' extrema do gene são alternativamente utilizados e não parecem contribuir para a sequência de codificação.32 A FBN1 está presente na pele, pulmões, rins, vasos sanguíneos, cartilagem, tendões, músculos, córnea e zônula ciliar. É constituída pela repetição de 47 domínios homólogos ao fator de crescimento epidérmico, 43 dos quais são domínios do fator de crescimento epidérmico ligado ao cálcio (cbEGF). Cada domínio contém seis (varia de seis a oito entre os estudos)32 resíduos de cisteína que formam três pontes de sulfeto, entre C1 e C3, C2 e C4, e C5 e C6, conferindo estabilidade à sua conformação tridimensional. Os domínios cbEGF têm um papel relevante na proteção contra a proteólise da FBN1, na promoção da interação entre os monômeros de FBN1 e os outros componentes das microfibrilas e na estabilização da estrutura das microfibrilas. A FBN1 contém também sete domínios homólogos47 aos encontrados nas proteínas latentes de ligação ao fator de crescimento beta‐1 (LTBPs) e ainda um módulo híbrido com características dos domínios cbEGF e LTBPs.1
Figura 5 - Localização do gene FBN1 no cromossomo 15

Fonte: Criado por Barbara Inácio da Silva 40
O gene da fibrilina-2 é homólogo ao da fibrilina-1, mutações nestes genes causam desordens fenotípicamente semelhantes. Alguns autores sugerem que a expressão do gene FBN2 direciona a montagem das fibras elásticas durante a embriogênese e a do gene FBN1 apresenta uma função estrutural maior das microfibrilas.33
Em 90% dos casos a Síndrome de Marfan resulta de mutações do gene da fibrilina‐1.34 Aproximadamente 25% dos casos resultam de uma mutação de novo (Figura 6), sendo que os restantes 75% herdam a condição de um parente afetado. O tipo de mutação do gene FBN1 mais frequentemente encontrado é a mutação pontual.1 Como regra geral, uma variante que causa a perda ou ganho in-frame da sequência de codificação central por meio de deleções, inserções ou erros de splicing47 está associada a doenças mais graves.33
Figura 6 - Mutação de novo

Fonte: Editado por Rafaela Campos. Disponível na US National Library of Medicine 41
Hereditariedade
A síndrome de Marfan, por ser uma herança autossômica dominante (Figura 7), deve afetar todas as gerações em uma família (Figura 8), ocorrer com a mesma frequência em homens e mulheres e o indivíduo afetado pode ser homo ou heterozigoto (mais comum).29 Isso significa que se um dos pais tiver a doença, ou seja, um gene normal e um gene mutado, a probabilidade de ambos os filhos contraírem a doença é de 50%.21 As crianças que não receberam o gene mutado não contraem a doença e também não a transmitem. A mutação geralmente ocorre em um dos gametas dos pais (óvulos ou espermatozoides) e a mutação de novo na criança torna-se hereditária e pode ser passada para a próxima geração.36
A incidência de diagnósticos esporádicos, no entanto, é de apenas 30%, enquanto que a incidência de Síndrome de Marfan entre pessoas de uma mesma família corresponde a todo o restante.35
Figura 7 - Herança autossômica dominante

Fonte: Editado por Vitória Gomes da Silva. Disponível na US National Library of Medicine 42
Figura 8 -Heredograma da Síndrome de Marfan

Fonte: Criado por Rafaela Campos43
Risco para membros da família
- É apropriado avaliar ambos os pais quanto a manifestações da síndrome de Marfan, realizando um exame clínico abrangente e ecocardiograma. Se a variante patogênica FBN1 foi identificada no probando, o teste genético molecular para esclarecer o status genético dos pais é possível.
- Se a variante patogênica FBN1 encontrada no probando não pode ser detectada no DNA de leucócitos de nenhum dos pais, as possíveis explicações incluem uma variante patogênica de novo no probando ou mosaicismo da linha germinativa em um dos pais. O mosaicismo da linha germinativa foi relatado em casos raros.
- Embora 75% dos indivíduos com diagnóstico de síndrome de Marfan tenham um dos pais afetados, a história familiar pode parecer negativa devido à falha em reconhecer o distúrbio em membros da família ou à morte prematura do pai antes do início dos sintomas.
- Quando os pais não são clinicamente afetados, o risco para os irmãos de um probando parece ser baixo, mas acima do risco populacional devido a casos relatados (mas raros) de mosaicismo somático e germinativo.32
Penetrância
A penetrância na SMF, que é a probabilidade de um gene ter qualquer expressão fenotípica, é praticamente 100%, significando que todo o indivíduo que herdar o gene mutante apresentará a doença. Apesar disso, a SMF tem uma ampla variação de intensidade clínica, apresentando uma expressividade variável, que pode estar associada a fatores ambientais e epigenéticos47. A expressividade é o grau de apresentação do fenótipo47. O número, idade de início e a constelação de manifestações variam enormemente entre os indivíduos afetados, mesmo dentro da mesma família. As mutações na FBN1 estão associadas com uma variação fenotípica alta, abrangendo desde manifestações isoladas de SMF à apresentação neonatal de doença severa e rapidamente progressiva em múltiplos órgãos.29
Pleiotropia
Um defeito congênito47 demonstra pleiotropia quando um único agente causador assinalado resulta em anormalidades de mais de um órgão em diferentes partes do embrião ou em múltiplas estruturas que surgem em diferentes momentos durante a vida intra-uterina. O agente responsável pela malformação pode ser também um gene mutante ou um teratógeno. Quando o agente causador causa múltiplas anormalidades em paralelo, o conjunto de anormalidades é denominado síndrome.7 Na Síndrome de Marfan, o gene FBN1 que codifica a proteína Fibrilina-1 sofre mutação e traz efeitos múltiplos afetando por exemplo: o tecido conjuntivo, coração, pulmões, ossos e olhos. Existem mais de 1.000 mutações diferentes descritas no FBN1 e elas causam não apenas a síndrome de Marfan, mas também outros tipos de doenças do tecido conjuntivo.21
Aconselhamento Genético
- O momento ideal para determinação do risco genético e discussão sobre a disponibilidadede testes pré-natais é antes da gravidez.
- É apropriado oferecer aconselhamento genético (incluindo discussão sobre os riscos potenciais para a prole e as opções reprodutivas) para os jovens adultos afetados.32
O banco de DNA é o armazenamento de DNA (normalmente extraído de células brancas do sangue) para possível uso futuro. Como é provável que a metodologia de teste e nossa compreensão dos genes, variantes alélicas e doenças melhorem no futuro, deve-se levar em consideração os bancos de DNA dos indivíduos afetados.32
Tratamento
O tratamento padrão inclui β‐bloqueantes profiláticos para diminuir a taxa de dilatação da aorta ascendente e cirurgia profilática da aorta. É importante ressaltar que a terapia com β- bloqueadores pode reduzir a ativação do TGF- β, que foi reconhecida como um fator contribuinte na síndrome de Marfan. O sucesso do tratamento médico e cirúrgico atual da patologia aórtica da síndrome de Marfan melhorou substancialmente a esperança média de vida, entre 1972 e 1993, a previsão de sobrevida em 50% dos pacientes subiu de 49 para 74 anos nas mulheres e de 41 para 70 anos nos homens.1 É imprescindível um acompanhamento multidisciplinar.
- Papel da fisioterapia em pacientes com a patologia
O caráter multissistêmico da Síndrome de Marfan requer uma abordagem multidisciplinar, que deve incluir a avaliação por especialistas em cardiologia, cirurgia cardiotorácica, ortopedia, reumatologia, oftalmologia e genética.8 O tratamento fisioterapêutico é parte integrante dessa equipe interdisciplinar que visa o auxílio terapêutico nos pacientes portadores da Síndrome de Marfan. A atuação deve ser considerada de fundamental importância uma vez que possui atuação em diversos sistemas tais como locomotor, cardiovascular e respiratório.
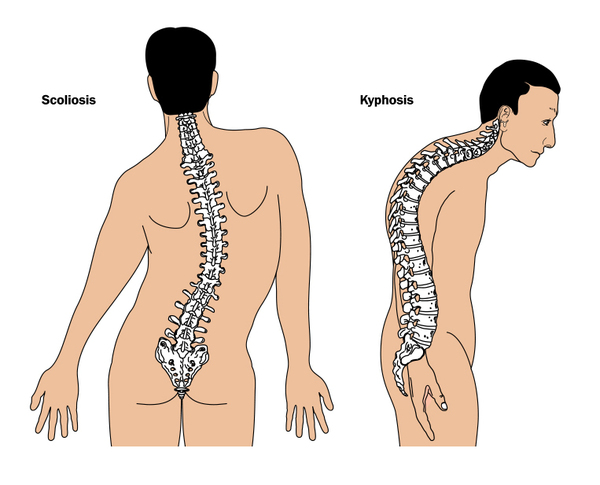
A atuação quanto ao aparelho locomotor baseia-se em uma terapêutica que tem como objetivo minimizar as deformidades esperadas nestes pacientes tais como cifose, escoliose (Figura 9), alteração da caixa torácica e aracnodactilia, que geram maior probabilidade de desenvolvimento de doenças respiratórias restritivas. O acompanhamento fisioterapêutico é de suma importância visando a manutenção dos volumes e capacidades pulmonares.A hipermobilidade das articulações faz com que estes indivíduos sejam mais propensos a desenvolver lesões e luxações, situação está que também deve ser associada ao processo preventivo e curativo com a terapêutica fisioterápica.23
Figura 9 - Escoliose e cifose

Fonte: Editado por Vitória Gomes da Silva. Disponível em: www.blamb/Shutterstock.com 44
A avaliação postural de pacientes com SM pode identificar precocemente o surgimento dessa deformidade antes que os sintomas apareçam. Nos casos mais severos de pacientes com escoliose de moderada a severa, podem-se observar complicações cardiorrespiratórias pela limitação da expansibilidade da caixa torácica devido ao desvio da coluna vertebral. O ângulo de Cobb é determinante para indicação do tratamento. Os pacientes com SM com escoliose não-estruturada apresentam boa evolução com o tratamento conservador, incluindo fisioterapia com ou sem associação de colete, que pode ser usado para estabilizar a coluna, especialmente durante os anos de formação. Se o tratamento não for eficaz e o desvio da coluna se tornar muito grande, a cirurgia pode ser necessária. Para aliviar os sintomas que se seguem aos pés chatos, é necessário um tratamento precoce na forma de palmilhas e sapatos adaptados individualmente.22
A fisioterapia respiratória, no pré e pós-operatório do paciente com SM, contribui para o sucesso no desmame ventilatório, diminuindo as disfunções respiratórias e, consequentemente, as complicações. A reabilitação pulmonar consiste em uma série de atividades e terapias como aconselhamento nutricional, técnicas de conservação de energia e estratégias de respiração.24
No sistema cardiovascular a fisioterapia exerce papel no que diz respeito à melhora do condicionamento cardiorrespiratório e musculoesquelético, através da realização de exercícios planejados e sob monitorização contínua, uma vez que estes pacientes podem ser classificados como indivíduos de risco. O paciente com Marfan só pode ser liberado para realização dessas atividades físicas após avaliação de um médico cardiologista. Feito isto, o Fisioterapeuta é adequado para prescrever e orientar o tipo de atividade a ser realizada, identificando características passíveis de prevenção e tratando as já instaladas.
Especificamente, o paciente deve ser educado sobre o risco para o sistema cardiovascular com esportes extenuantes e de contato e deve ser informado sobre as Diretrizes de atividade física da National Marfan Foundation, preparadas pelo NMF Professional Advisory Board.28 Como o tecido conjuntivo desses pacientes é mais fraco do que o normal, existem exercícios apropriados para evitar problemas indesejados. As atividades não devem apresentar caráter competitivo, exercícios isométricos (realização de força sem movimento), como por exemplo musculação, não são benéficos, estando mais indicados os exercícios isotônicos (realização de força com movimento, como por exemplo caminhadas) com intensidade leve à no máximo moderada. Devem ser evitadas mudanças bruscas de direção, paradas abruptas ou colisões com outros jogadores ou equipamentos e quedas no chão.25 O exercício promove não só uma melhora da capacidade física, como também tem efeito benéfico sobre o controle da pressão arterial e da densidade óssea. Exercícios físicos apropriados e seguros são importantes para aumentar tanto a sobrevida quanto a qualidade de vida.
O fisioterapeuta pode atuar, também, ajudando no remodelamento cardíaco que é definido como alterações reversíveis ou não das estruturas e dos fenômenos bioquímicos dos compartimentos musculares e/ou intersticiais47 do miocárdio, sendo de caráter adaptativo, porém seguido de deterioração funcional.29
Glossário47
Alelos: são as formas alternativas de um determinado gene e ocupam um mesmo loco em cromossomos homólogos.
Aneurisma da aorta proximal: um alargamento da aorta, o principal vaso sanguíneo que fornece sangue ao corpo, no nível do abdômen
Bolhas apicais: bolhas pulmonares são achadas radiológicos frequentes em pulmões dos portadores de enfisema pulmonar
Congênito: característico do indivíduo desde o início ou antes do nascimento.
Delgados: finos, magros.
Dissecção aórtica: dissecção aórtica, é a parte da aorta que atravessa o tórax. Quando o revestimento da aorta se rompe, o sangue pode penetrar pela ruptura e separar (dissecar) a camada média da parede da aorta da camada externa, ainda intacta.
Dolicocefalia: é um tipo de craniosynostosis , em que não é um fecho prematuro da sutura sagital exclusiva ou interparietal do crânio, que liga os dois ossos parietais.
Domínios homólogos: proteínas da mesma família.
Ectasia dural lombossacra: o alargamento do saco dural em torno da coluna é um achado comum e ocorre mais frequentemente na espinha lombossacra.
Epigenética: é a área da biologia que estuda mudanças no funcionamento de um corpo que não são causadas por momentos na sequência de DNA e que se perpetuam nas divisões celulares, meióticas ou mitóticas.
Estrabismo: distúrbio em que os olhos não olham exatamente na mesma direção ao mesmo tempo.
Estria atrófica: alterações cutâneas caracterizadas pela perda de colágeno e elastina na derme, assemelhando-se a cicatrizes.
Exons: são sequências de nucleotídeos que serão conservadas na criação do RNA maduro. São transcritos e traduzidos.
Fenótipo: refere-se às características visíveis que podem ser modificadas.
Genótipo: é o conjunto formado pelos genes de um indivíduo que não são modificados naturalmente.
Glicoproteína: proteínas que contêm glicanos anexadas a cadeias laterais de polipeptídios.
Hérnia: é a protusão, ou seja, o escape parcial ou total de um ou mais órgãos por um orifício que se abriu, por má formação ou enfraquecimento, nas camadas de tecido protetoras dos órgãos internos do abdômen.
Intersticial: substância sólida ou líquida que existe entre as células de quase todos os tecidos animais, e que se denomina também substância fundamental.
Laxidez: fadiga.
Luxação: perda da congruência articular, ou seja, quando um osso sai por completo de sua posição anatômica correta.
Monômeros: molécula que forma uma unidade básica de polímeros.
Multissistêmica: afeta vários sistemas do corpo.
Palato ogival: com a respiração pela boca, a língua não fica em contato, ocasionando um crescimento anormal e mais estreito do palato
Pneumotórax: é uma urgência médica relativamente comum, que é causada pela entrada de ar dentro da pleura, a membrana que recobre os pulmões.
Predileção: preferência acentuada por algo.
Probando: Termo que designa o indivíduo que está sendo estudado.
Retrognatismo: um tipo de má oclusão maxilar causada pela posição mais posterior da mandíbula.
Seios de Valsalva: Refere-se às três bolsas atrás das válvulas do coração.
Splicing: processo de maturação de um pré-mRNA (RNA precursor), nesse processo as regiões não codificantes (íntrons) são retiradas do pré-mRNA, que passa a conter somente as regiões codificantes (éxons).
Válvulas auriculoventriculares: Consiste na separação dos átrios (aurículas), esquerdo e direito de seus respectivos ventrículos.
Vítreo: É uma substância gelatinosa e viscosa que se encontra no segmento posterior do olho, entre o cristalino e a retina.
- Produto produzido sobre Síndrome de marfan:
O produto está em dois formatos. Trata-se de um mangá com o podcast narrando as ilustrações do Mangá.
Esse é o link para as ilustrações do Mangá sobre Síndrome de Marfan.
Agora acesse esse link para o podcast narrando a história do Mangá. Abra os dois links para ter uma melhor experiência.
- Equipe:




Bárbara Inácio Isabela de Paiva Vitória Gomes Rafaela Campos
da Silva Locatelli da Silva
- REFERÊNCIAS
- Coelho SG, Almeida AG. Marfan syndrome revisited: From genetics to the clinic. Rev Port Cardiol. 2020 Apr;39(4):215-226. English, Portuguese. doi: 10.1016/j.repc.2019.09.008. Epub 2020 May 18. PMID: 32439107. Acesso em: 25 nov. 2020.
2. ORPHA NET. Síndrome de Marfan. Disponível em: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=pt&Expert=558. Acesso em: 27 out. 2020.
3. Judge DP, Dietz HC. Marfan's syndrome. Lancet. 2005 Dec 3;366(9501):1965-76. doi: 10.1016/S0140-6736(05)67789-6. PMID: 16325700; PMCID: PMC1513064. Acesso em: 25 set. 2020.
4. Salik I, Rawla P. Marfan Syndrome. 2020 Jun 24. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan–. PMID: 30726024. Acesso em: 25 set. 2020.
5. Wagner AH, Zaradzki M, Arif R, Remes A, Müller OJ, Kallenbach K. Marfan syndrome: A therapeutic challenge for long-term care. Biochem Pharmacol. 2019 Jun;164:53-63. doi: 10.1016/j.bcp.2019.03.034. Epub 2019 Mar 27. PMID: 30926475. Acesso em: 25 set. 2020.
6. Yuan SM, Jing H. Marfan's syndrome: an overview. Sao Paulo Med J. 2010 Dec;128(6):360-6. doi: 10.1590/s1516-31802010000600009. PMID: 21308160. Acesso em: 25 set. 2020.
7. Robert L. Nussbaum, et al. THOMPSON & THOMPSON GENÉTICA MÉDICA. 7ª Edição ed., Elsevier Editora Lt da, 2008.
8. McKusick, V. A. Heritable Disorders of Connective Tissue. (4th ed.) St. Louis: C. V. Mosby (pub.) 1972. Acesso em: 02 dez. 2020
9. Pyeritz, R. E., McKusick, V. A. The Marfan syndrome. New Eng. J. Med. 300: 772-777, 1979. Acesso em: 02 dez. 2020
10. Pyeritz, R. E. The Marfan syndrome. In: Royce, P. M.; Steinmann, B.: Connective Tissue and Its Heritable Disorders: Molecular, Genetic, and Medical Aspects. New York: Wiley-Liss (pub.) 1993. Pp. 437-468. Acesso em: 02 dez. 2020
11. GENOME. About Marfan Syndrome. Disponível em: https://www.genome.gov/Genetic-Disorders/Marfan-Syndrome. Acesso em: 27 out. 2020.
12. Eaton L, Meiner SE. Marfan syndrome: identification and management. Medsurg Nurs. 1999 Apr;8(2):113-7. PMID: 10410009. Acesso em: 02 dez. 2020
13. Patrocinador, PD, Hobbs, W., Riley, LH, III, Pyeritz, RE A coluna toracolombar na síndrome de Marfan. J. Bone Joint Surg. Sou. 77: 867-876, 1995. Acesso em: 02 dez. 2020
14. SCIELO. Síndrome de Marfan. Disponível em: http://www.scielo.mec.pt/scielo.php?script=sci_arttext&pid=S0872-07542016000400002&lng=pt&nrm=iso. Acesso em: 29 out. 2020.
15. Kumar A, Agarwal S. Marfan syndrome: An eyesight of syndrome. Meta Gene. 2014 Jan 14; 2:96-105. doi: 10.1016/j.mgene.2013.10.008. PMID: 25606393; PMCID: PMC4287801. Acesso em: 02 dez. 2020
16. De Backer J. Cardiovascular characteristics in Marfan syndrome and their relation to the genotype. Verh K Acad Geneeskd Belg. 2009;71(6):335-71. PMID: 20232788. Acesso em: 02 dez. 2020
17. TESES USP. Análise de ligação na Síndrome de Marfan. Disponível em: https://www.teses.usp.br/teses/disponiveis/87/87131/tde-30042010-084608/publico/LuciaValeriaSilvaTeixeira_Doutorado.pdf. Acesso em: 27 out. 2020.
18. INTERNETMEDICA. Marfan Syndrome. Disponível em: https://internetmedica.com.br/sindrome-de-marfan-online-marfan-syndrome/. Acesso em: 27 out. 2020.
19. Lundby R, Rand-Hendriksen S, Hald JK, Lilleås FG, Pripp AH, Skaar S, Paus B, Geiran O, Smith HJ. Dural ectasia in Marfan syndrome: a case control study. AJNR Am J Neuroradiol. 2009 Sep;30(8):1534-40. doi: 10.3174/ajnr.A1620. Epub 2009 May 20. PMID: 19461064; PMCID: PMC7051621. Acesso em: 12 de novembro de 2020
20. Izquierdo NJ, Traboulsi EI, Enger C, Maumenee IH. Strabismus in the Marfan syndrome. Am J Ophthalmol. 1994 May 15;117(5):632-5. doi: 10.1016/s0002-9394(14)70069-8. PMID: 8172269. Acesso em: 27 out. 2020.
21. SOCIAL STYRELSEN. Marfans syndrom. Disponível em: https://www.socialstyrelsen.se/stod-i-arbetet/sallsynta-halsotillstand/marfans-syndrom/. Acesso em: 29 out. 2020.
22. The Marfan Foundation. A Guide to Marfan Syndrome and Related Disorders. Disponível em: http://info.marfan.org/hubfs/Resource_Downloads/Marfan_Syndrome_and_Related_Disorders.pdf Acesso em: 12 nov. 2020
23. Yolanda Smith. “Tratamento da síndrome de Marfan.” 23 agosto 2018, https://www.news-medical.net/health/Marfan-Syndrome-Treatment-(Portuguese).aspx. Acesso em: 12 nov. 2020.
24. Paulo Peres, and Gabriella França B. Cipriano. “MARFAN Grupo Multidisciplinar Síndrome de Marfan.” Exercício e Síndrome de Marfan, http://www.marfan.com.br/vivendo_marfan/fisioterapia/images/folheto_marfan.pdf. Acesso em: 12 nov. 2020.
25. Robert L. Nussbaum, et al. THOMPSON & THOMPSON
26. SCIELO. Avaliação antropométrica e musculoesquelética de pacientes com síndrome de Marfan. Disponível em: https://www.scielo.br/scielo.php?script=sci_arttext&pid=S1413-35552011000400006&lng=pt&nrm=iso&tlng=pt. Acesso em: 12 nov. 2020.
27. PHYSIOPEDIA. Marfan Syndrome. Disponível em: https://www.physio-pedia.com/Marfan_Syndrome#:~:text=Physical%20Therapy%20Management&text=Stretching%2C%20strengthening%2C%20therapeutic%20exercises%2C,the%20patient%20with%20Marfan%20syndrome. Acesso em: 12 nov. 2020.
28. Medeiros, Wladimir Musetti, Peres, Paulo Alberto, Carvalho, Antônio Carlos, Gun, Carlos, & De Luca, Fábio Augusto. (2012). Efeito de um programa de exercício físico em portador da Síndrome Marfan com disfunção ventricular. Arquivos Brasileiros de Cardiologia, 98(4), e70-e73. https://doi.org/10.1590/S0066-782X2012000400015. Disponível em: https://www.scielo.br/pdf/abc/v98n4/v98n4a15.pdf Acesso em: 12 nov. 2020
29. MAURÍCIO MENNA BARRETO, et al. “SÍNDROME DE MARFAN.” novembro de 2002, http://www.marfan.com.br/pesquisas_tratamento/trabalhos/trabalhos_cientificos/sindrome_marfan.pdf. Acesso em: 02 dezembro 2020.
30. Sakai LY, Keene DR. Fibrillin protein pleiotropy: Acromelic dysplasias. Matrix Biol. 2019 Jul;80:6-13. doi: 10.1016/j.matbio.2018.09.005. Epub 2018 Sep 13. PMID: 30219651. Acesso em: 02 nov. 2020. Disponível em: https://pubmed.ncbi.nlm.nih.gov/30219651/
31. ANA LEBREIRO, et al. “Síndrome de Marfan: manifestações clínicas, fisiopatologia e novas perspectivas da terapêutica farmacológica.” vol. 29, Junho 10. Acesso em: 02 de dezembro de 2020
32. NCBI. Marfan Syndrome. Disponível em: https://www.ncbi.nlm.nih.gov/books/NBK1335/. Acesso em: 2 dez. 2020.
33. SCIELO. Anomalias oculares e características genéticas na síndrome de Marfan. Disponível em: https://www.scielo.br/scielo.php?script=sci_arttext&pid=S0004-27492002000600005#:~:text=Trata%2Dse%20de%20uma%20doen%C3%A7a,S%C3%ADndrome%20de%20Marfan(1). Acesso em: 18 nov. 2020
34. PUBMED. Síndrome de Marfan revisitada: da genética à clínica. Disponível em: https://pubmed.ncbi.nlm.nih.gov/32439107/ . Acesso em: 1 dez. 2020
35. MINHA VIDA. Síndrome de Marfan: sintomas, tratamentos e causas. Disponível em: https://www.minhavida.com.br/saude/temas/sindrome-de-marfan#:~:text=A%20incid%C3%AAncia%20de%20diagn%C3%B3sticos%20espor%C3%A1dicos,a%20doen%C3%A7a%20para%20os%20filhos. Acesso em: 2 dez. 2020.
36. ONCOGUIA. Alterações nos Genes. Disponivel em: http://www.oncoguia.org.br/conteudo/alteracoes-nos-genes/8160/73/ Acesso em: 2 dez. 2020.
37. Figura 2 - Medline Plus. Disponível em: https://www.ncbi.nlm.nih.gov/books/NBK148668/figure/mps4a.F4/?report=objectonly. Acesso em: 04 jan. 2020.
38. Figura 3 - Medline Plus. Disponível em: https://medlineplus.gov/images/PX0001AW_PRESENTATION.jpeg. Acesso em: 04 jan. 2020.
39. Figura 4 - Medline Plus. Disponível em: https://medlineplus.gov/images/PX0002MI_PRESENTATION.jpeg. Acesso em: 04 jan. 2020.
40. Figura 5 - Criado por Barbara Inácio da Silva.
41. Figura 6 - Medline Plus. Disponível em: https://medlineplus.gov/images/PX0000A8_PRESENTATION.jpeg. Acesso em: 04 jan. 2020.
42. Figura 7 - Medline Plus. Disponível em: https://medlineplus.gov/images/PX00009C_PRESENTATION.jpeg. Acesso em: 04 jan. 2020.
43. Figura 8 - Criado por Rafaela Campos
44. Figura 9 - Medline Plus. Disponível em: https://medlineplus.gov/images/PX0001AC_PRESENTATION.jpeg. Acesso em: 04 jan. 2020.
45. Figura 1 - Byers PH. Determination of the molecular basis of Marfan syndrome: a growth industry. J Clin Invest. 2004 Jul;114(2):161-3. doi: 10.1172/JCI22399. PMID: 15254580; PMCID: PMC449756. Acesso em: 06 jan. 2021
46. Quadro 1 - Disponível em: https://www.teses.usp.br/teses/disponiveis/87/87131/tde-30042010-084608/publico/LuciaValeriaSilvaTeixeira_Doutorado.pdf. Acesso em: 06 jan. 2021
47. Glossário
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}